Sequence Search:
Sequence search is a method of searching similarities between a nucleotide/protein sequence and a query string from sequence databases by using alignment and identifying how much percentage of similarity is present between the query and subject sequence using sequence alignment software tools such as BLAST, Clustal, MAFFT, etc.
What is DNA sequencing and how it is done?
DNA sequencing is the process of determining the nucleotide (adenine, cytosine, guanine, and thymine) order of a given DNA fragment. Sequencing plays a vital role in identifying changes in genes and noncoding DNA (including regulatory sequences), associations with diseases and phenotypes, and identify potential drug targets. It has become a key technology in many areas including evolutionary biology and forensics.
Most DNA sequencing has been performed using the chain termination method developed by Frederick Sanger called Sanger sequencing which is based on random incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication.
Next-generation sequencing (NGS) is a technology which is used to sequence the orientation of nucleotides in genetic material. It is also called as massively parallel sequencing which use miniaturized and parallelized platforms for sequencing of 1 million to 43 billion short reads (50 to 400 bases each) per instrument run. This technology enables the sequencing of DNA or RNA more quickly and at a lower cost than Sanger sequencing. especially for large-scale, automated genome analyses
Next Generation Sequencing (NGS) market is expected to reach $27.5 billion by 2030, at a CAGR (Compound Annual Growth Rate) of 15.8%, says Meticulous Research®.
Pairwise to Multiple sequence alignment
Pairwise sequence alignments are calculated by BLAST, FASTA, and SSEARCH which is used to view the evolutionary structure of a protein or domain family from a single perspective. Pairwise alignments produce very accurate statistical significance estimates and perfect homology are found. Searches with models of protein families, using either PSI-BLAST or Hidden Markov Model (HMM) based methods, can identify far more homologs in a single search at little additional computational cost. Moreover, the multiple sequence alignments that are used to construct the position-specific scoring matrices of PSI-BLAST or the Hidden Markov Models used by HMMER (software package for sequence analysis which is used to identify homologous protein or nucleotide sequences, and to perform sequence alignments) provide important information about the most conserved regions in the protein; locating conserved regions in protein and domain families can dramatically improve predictions of the functional consequences of mutation. Multiple sequence alignments thus provide much more structural, functional, and phylogenetic information than pairwise alignments.
While multiple sequence alignments are much more informative, they cannot be used to find out the homology of two sequences which is more important criteria in sequence mapping. The inclusion of a protein into a multiple sequence alignment requires independent evidence for homology; multiple sequence alignment programs do not provide statistical estimates and will readily align non-homologous sequences, particularly non-homologous sequences that are highly ranked by chance in a similarity search. The assumption of homology can be especially misleading with iterative methods like PSI-BLAST, because once a non-homologous domain has been included in the multiple sequence alignment used to produce the position-specific scoring matrix, the matrix can become re-purposed towards finding members of the non-homologous family. Ideally, each sequence included in a multiple sequence alignment will be evaluated both to ensure that it shares significant similarity with some of the other members of the family, and that the boundaries of the included sequence correspond with the boundaries of the domain homology.
Meticulously building a multiple sequence alignment is exponentially more computationally expensive than pairwise alignment. Exact pairwise alignment algorithms require time proportional to the product of two sequences, so increasing the sequence lengths 2-fold increased the time required 4-fold. Fortunately, protein sequences have a limited range of lengths, so rigorous searches (SSEARCH) are routine. In contrast, the rigorous multiple sequence alignment of 10 sequences of length 400 would take proportional to 40010, so rigorous multiple sequence alignment is impractical. During the 1980s, progressive alignment strategies, like ClustalW were developed that simplified the problem to O(n2l2), where n is the number of sequences, and l is their average length. These early progressive alignment strategies suffered from the problem that gaps placed early on in the alignment could not be re-adjusted to reflect information from sequences aligned later. More recent multiple sequence alignment methods, like MAFFT and MUSCLE use iterative approaches that allow gaps to be re-positioned.
There is a much greater diversity of multiple sequence alignment algorithms than pairwise sequence alignment algorithms, largely because optimal pairwise solutions are readily available, but multiple sequence alignment strategies use different heuristic approximations. While different multiple sequence alignment programs will often produce modestly different results, most programs produce very similar results for sequences at modest evolutionary distances (greater than 40% identity), and the differences are found near the boundaries of gaps. A more common problem is multiple sequence alignments with large gaps, which may reflect the presence or absence of domains in a subset of the sequence set. Alignment only makes biological sense when the residues included in the alignment are homologous. Large differences in sequence length, or attempts to multiply align sequences that are locally, but not globally, homologous can produce very different results because the programs are aligning non-homologous domains.
Databases to perform Sequence search
Sequence search is performed to identify a specified sequence of amino acids/ genes/ peptides, etc. in patent or non-patent literatures. It is used to check whether the bio sequence is novel because the novelty of the bio sequence is dependent on the distinctive combination of nucleotides or amino acids. Keyword based search alone will not be effective to extract the entire relevant data set, hence a thorough sequence search is a must.
Various smart bio sequence databases STNext, CAS registry, MMS Gene, Genome Quest, DCR, NCBI Blast, Derwent SequenceBase, Patent Lens, Orbit Biosequence, etc. are used to determine all relevant prior arts particularly on protein and nucleotide sequences. These databases will give results based on sequence similarity / identity and align the query sequence to the sequence present in the patent / NPL / subject sequence. Sequence similarity search can also be performed on BLAST, FASTA, HMMER, iProClass, and specifically for searching protein sequences InterProScan, ScanProsite can be used.
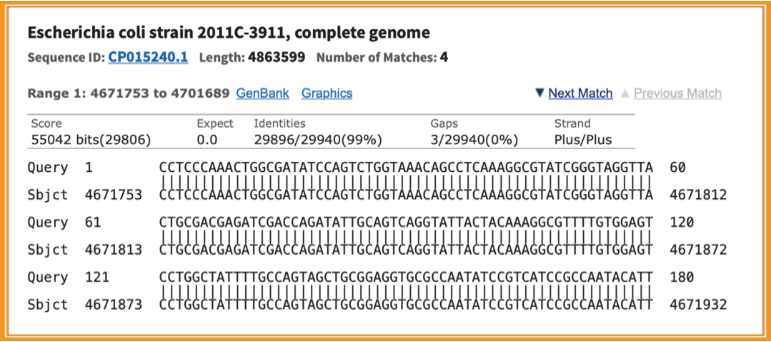
Below is the sequence alignment of the query sequence vs. the NCBI nucleotide database sequences which includes the Karlin-Altschul Expect value, the percent identity of the alignment, the number of gaps in the alignment, and other information.
How can MCRPL help?
MCRPL is a 20-year-old company that undertakes all type of searches involving all scientific, and technical information. Our team consists of several experts holding Masters and Ph.D. degrees in different fields. The team is highly experienced in sequence searches and sequence listing, with an avg. experience of more than 4 years in this specialization. We apply extensive resources and knowledge to work on several challenging and extremely complex searches.
We use the Human-plus-Machine approach (MCRANK) to ensure that our quality output is immaculate, with well-maintained and documented SOPs, and internal manuals for working on typical IP projects. We have many patent agents and patent attorneys that give us an edge in performing complex searches.
© Molecular Connections Private Limited
For more information, contact priorart@molecularconnections.com
For more updates subscribe IP Tech Insider
Also, you can place an order for your search on our online portal: https://ipsolutionshub.com/




